1. a-Талассемия - это гемолитическая анемия вследствие дефицита синтеза a глобина в результате потери или повреждения одного или нескольких a-глобиновых генов. Снижение синтеза a цепей приводит к накоплению свободных g- и b-глобиновых цепей и формированию из них тетрамеров - g4 (Hb Bart's) и нестабильного b4 (Hb H), с последующим ускорением разрушения эритроцитов. Эти тетрамеры обладают очень высоким сродством к кислороду и не могут выполнять функцию переноса кислорода. Клиническая картина тяжелой a-талассемии является следствием комбинации гипохромной анемии, гемолиза и дефектного транспорта кислорода по причине различных количеств физиологически неэффективного Hb в эритроцитах. В результате, степень тканевой гипоксии значительно больше, чем можно ожидать от имеющейся степени анемии.
Клинически a-талассемия делится на четыре группы: "немое" носительство, a-талассемия с минимальными изменениями, гемоглобинопатия Н (HbH), a-талассемия с водянкой плода. Тяжесть фенотипического проявления a-талассемии прямо пропорциональна снижению a-глобинового синтеза.
- "Немое" носительство - потеря одного a-глобинового гена ( ). Фенотипически "немые" носители a-тал трудно отличимы от здоровых. MCV обычно в пределах 78 - 80 фл при этом МСН может быть на нижней границе нормы. Все другие гематологические параметры соответствуют референтным значениям. Некоторые "немые" носители могут иметь даже нормальные показатели MCV в пределах 80 - 85 фл. В периоде новорожденности некоторые из них имеют небольшие количества Hb Bart's в крови (менее 2%), которое исчезает в течение первых месяцев жизни.
). Фенотипически "немые" носители a-тал трудно отличимы от здоровых. MCV обычно в пределах 78 - 80 фл при этом МСН может быть на нижней границе нормы. Все другие гематологические параметры соответствуют референтным значениям. Некоторые "немые" носители могут иметь даже нормальные показатели MCV в пределах 80 - 85 фл. В периоде новорожденности некоторые из них имеют небольшие количества Hb Bart's в крови (менее 2%), которое исчезает в течение первых месяцев жизни.
- a-Талассемия с минимальными изменениями - заболевание, развивающееся при потере двух a-глобиновых генов (![]() ,
, ![]() ,
, 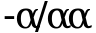 и
и ![]() ). Эти пациенты как правило не имеют клинических проявлений. Лабораторные исследования их крови выявляют микроцитоз с MCV в пределах 70 - 75 фл, гипохромию с МСН в пределах 18 - 22 пг, повышение числа эритроцитов, дисбаланс в a/b биосинтетического соотношения (около 0,6), относительное содержание Hb A2 на нижней границе нормы. Некоторые, особенно дети, могут иметь легкую анемию, что при наличии легкого микроцитоза с гипохромией часто приводит к ошибочному диагнозу железодефицитной анемии. Все новорожденные при этом состоянии имеют 5 - 10% Hb Bart's в крови, который исчезает в течение первых месяцев жизни.
). Эти пациенты как правило не имеют клинических проявлений. Лабораторные исследования их крови выявляют микроцитоз с MCV в пределах 70 - 75 фл, гипохромию с МСН в пределах 18 - 22 пг, повышение числа эритроцитов, дисбаланс в a/b биосинтетического соотношения (около 0,6), относительное содержание Hb A2 на нижней границе нормы. Некоторые, особенно дети, могут иметь легкую анемию, что при наличии легкого микроцитоза с гипохромией часто приводит к ошибочному диагнозу железодефицитной анемии. Все новорожденные при этом состоянии имеют 5 - 10% Hb Bart's в крови, который исчезает в течение первых месяцев жизни.
- Гемоглобинопатия Н - средней тяжести гемолитическое заболевание, сопровождающееся повышенной усталостью, общим дискомфортом, спленомегалией, является результатом потери трех из четырех a-глобиновых генов (![]() ,
, ![]() , или
, или  ). Тяжесть этого состояния может значительно варьировать. Большинство пациентов (генотип
). Тяжесть этого состояния может значительно варьировать. Большинство пациентов (генотип ![]() или
или ![]() ) относятся к ТНЗТ и нуждаются в заместительных трансфузиях донорских эритроцитов в основном при интеркуррентных заболеваниях, оперативных вмешательствах или беременности. Остальные, с генотипом
) относятся к ТНЗТ и нуждаются в заместительных трансфузиях донорских эритроцитов в основном при интеркуррентных заболеваниях, оперативных вмешательствах или беременности. Остальные, с генотипом  , - трансфузионно зависимы с рождения. Гематологическими находками являются анемия от легкой до среднетяжелой степени с микроцитозом и гипохромией. Морфология эритроцитов значительно изменена - базофильная пунктация, изменение формы в виде мишеневидных и каплеобразных эритроцитов. При окраске бриллиантовым крезиловым синим видны преципитаты Hb Н в виде множественных точечных включений, которые превращаются в большие агрегаты после спленэктомии. In vitro биосинтез цепей демонстрирует значительное снижение a/b соотношения (~ 0,3).
, - трансфузионно зависимы с рождения. Гематологическими находками являются анемия от легкой до среднетяжелой степени с микроцитозом и гипохромией. Морфология эритроцитов значительно изменена - базофильная пунктация, изменение формы в виде мишеневидных и каплеобразных эритроцитов. При окраске бриллиантовым крезиловым синим видны преципитаты Hb Н в виде множественных точечных включений, которые превращаются в большие агрегаты после спленэктомии. In vitro биосинтез цепей демонстрирует значительное снижение a/b соотношения (~ 0,3).
- a-Талассемия с водянкой плода развивается при потере всех четырех a-глобиновых генов (--/--) является не совместимой с жизнью и приводит к мертворождению с водянкой плода. Основной Hb у этих плодов - Hb Bart's (g4). Остаточный синтез эмбриональной z-глобиновой цепи обеспечивает образование следового количества тетрамера Hb (Hb Portland) у плода в течение второй половины беременности. Более 50% новорожденных - мертворожденные на разных сроках беременности, оставшиеся погибают в первые часы жизни.
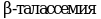
Три клинических синдрома связаны с различиями в степени тяжести и исторически названы большая форма b-талассемии, малая форма b-талассемии и промежуточная форма b-талассемия. Тяжесть клинической картины b-талассемии прямо пропорционально степени дисбаланса глобиновых цепей и зависит в основном от остаточной продукции b-глобиновых цепей. Два других фактора могут оказывать положительное влияние на дисбаланс цепей: повышение продукции g-глобиновых цепей и уменьшение синтеза a-глобиновых цепей вследствие сонаследования a-талассемии.
- Большая форма b-талассемии - типичные клинико-гематологические черты заболевания появляются на 3 - 6 месяце жизни, после переключения глобинового синтеза на взрослый тип, и проявляются в виде тяжелой гипохромной, микроцитарной, гемолитической анемии, требующих регулярных гемотрансфузий для поддержания адекватного Hb. У детей, не получающих заместительную терапию донорскими эритроцитами, тяжелая анемия приводит к ранней смертности в возрасте 3 - 4 лет (b0-тал) или в возрасте 8 - 12 лет (b+-тал). К клиническим проявлениям также относятся прогрессирующее увеличение печени и селезенки, типичные изменения плоских костей за счет экспансии костного мозга и патологические переломы трубчатых костей. Вторичный гиперспленизм, приводящий к тромбоцитопении, лейкопении и ускоренному разрушению трансфузированных эритроцитов, еще больше осложняет клиническую картину. Задержка роста и полового развития начинает обращать на себя внимание к 10 годам. Течение заболевания у неадекватно леченных больных осложняется перегрузкой железа в результате трансфузий и повышенной всасываемости железа в кишечнике. Гематологически большая форма b-талассемии имеет в дополнении к глубокой анемии типичные изменения морфологии эритроцитов в виде гипохромии, микроцитоза, анизопойкилоцитоза, полихромазии, фрагментации, базофильной пунктации, многочисленных нормобластов в периферической крови. Число ретикулоцитов обычно невысоко в виду массивной деструкции эритроидных предшественников в костном мозге. Костный мозг гиперклеточный, с выраженной гиперплазией эритроидного ростка с бедно гемоглобинизированными нормобластами.
Без заместительной терапии среднее содержание Hb составляет 50 - 60 г/л у гомозигот по b0-талассемии, и несколько выше - около 70 г/л у гомозигот по b+-талассемии. Относительное содержание Hb A2 варьирует в значительных пределах, Hb F всегда повышено и может составлять до 98% от общего Hb.
- Малая форма b-талассемии. Гетерозиготы по b+-талассемии как правило бессимптомны, Hb на нижней границе нормы, гетерозиготы по b0-талассемии имеют анемию 1 степени. Эритроцитарные индексы MCV и MCH снижены до типичного значения в 60 - 70 фл (норма 85 - 92 фл) и 20 - 25 пг (норма 27 - 32 пг), соответственно. Гематологические характеристики включают также микроцитоз, гипохромию, анизопойкилоцитоз с мишеневидностью и базофильной пунктацией эритроцитов периферической крови, и легкую эритроидную гиперплазию в костном мозге. Увеличение селезенки редко и обычно весьма незначительно. Диагностическим тестом является количественная оценка соотношения типов гемоглобина (фракции гемоглобина): Hb A2, который как правило в 2 раза выше нормы (норма 2 - 3,5%), Hb F слегка повышен в половине случаев до ![]() . In vitro биосинтез цепей глобина выявляет дефицит b цепей, a/b соотношение 1,5 - 2,0 (норма 0,9 - 1,1). Крайне редко гетерозиготы по b++-талассемии могут иметь нормальные значения Hb F, Hb A2, MCV и МСН.
. In vitro биосинтез цепей глобина выявляет дефицит b цепей, a/b соотношение 1,5 - 2,0 (норма 0,9 - 1,1). Крайне редко гетерозиготы по b++-талассемии могут иметь нормальные значения Hb F, Hb A2, MCV и МСН.
- Промежуточная форма b-талассемии - объединяет весь спектр клинических синдромов по тяжести между большой и малой формами b-талассемии. Пациенты с промежуточной формой b-талассемии обычно в состоянии поддерживать содержание Hb в диапазоне 65 - 90 г/л, имеют гепатоспленомегалию типичную "талассемическую" деформацию плоских костей скелета и могут обходиться без регулярных заместительной трансфузий донорских эритроцитов.
Гемоглобины с гибридными цепями относятся согласно классификации к группе количественных гемоглобинопатий, ярким представителем которой является Hb Lepore. В процессе негомологичного кроссинговера образуются 2 аномальные хромосомы: Lepore - хромосома с ~7 kb делецией, которая имеет db гибридный ген вместо нормальных d и b генов, и "анти-Lepore" - хромосома, имеющая нормальные d и b гены и сливной bd ген между ними. Гомозиготное состояние Hb Lepore может привести к средним или тяжелым трансфузионно-зависимым талассемическим синдромам.
Характеристика основных форм талассемий в зависимости от генетического дефекта представлены в таблице 1.
Таблица 1. Клинико-генетическая характеристика форм талассемий
 ,
, 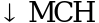
 ,
,
 ,
, , спленомегалия, HbH, часто требуются заместительные трансфузии
, спленомегалия, HbH, часто требуются заместительные трансфузии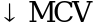 ,
,  , гепатоспленомегалия, Hb Bart, трансфузионная зависимость с внутриутробного периода
, гепатоспленомегалия, Hb Bart, трансфузионная зависимость с внутриутробного периода
 ;
;  ;
;  ;
; ;
;  ,
, , незначительная спленомегалия
, незначительная спленомегалия ;
; 
 ,
,  ,
, 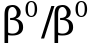 в сочетании с a-талассемией или с повышенным синтезом g-глобиновых цепей
в сочетании с a-талассемией или с повышенным синтезом g-глобиновых цепей ,
,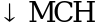 , значительная спленомегалия, гепатомегалия, часто требуются заместительные трансфузии
, значительная спленомегалия, гепатомегалия, часто требуются заместительные трансфузии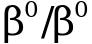 ,
,  ,
, 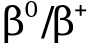
 ,
, , значительная спленомегалия, гепатомегалия;
, значительная спленомегалия, гепатомегалия;![]() - продукция
- продукция  цепи незначительно снижена;
цепи незначительно снижена; ![]() - продукция
- продукция  цепи значительно снижена;
цепи значительно снижена; ![]() - продукция
- продукция  полностью отсутствует.
полностью отсутствует.
- Гражданский кодекс (ГК РФ)
- Жилищный кодекс (ЖК РФ)
- Налоговый кодекс (НК РФ)
- Трудовой кодекс (ТК РФ)
- Уголовный кодекс (УК РФ)
- Бюджетный кодекс (БК РФ)
- Арбитражный процессуальный кодекс
- Конституция РФ
- Земельный кодекс (ЗК РФ)
- Лесной кодекс (ЛК РФ)
- Семейный кодекс (СК РФ)
- Уголовно-исполнительный кодекс
- Уголовно-процессуальный кодекс
- Производственный календарь на 2025 год
- МРОТ 2026
- ФЗ «О банкротстве»
- О защите прав потребителей (ЗОЗПП)
- Об исполнительном производстве
- О персональных данных
- О налогах на имущество физических лиц
- О средствах массовой информации
- Производственный календарь на 2026 год
- Федеральный закон "О полиции" N 3-ФЗ
- Расходы организации ПБУ 10/99
- Минимальный размер оплаты труда (МРОТ)
- Календарь бухгалтера на 2026 год
- Частичная мобилизация: обзор новостей
- Постановление Правительства РФ N 1875